
【诊断】从最新专家共识,谈中国法布雷病的诊疗
2024-06-12 网络 网络 发表于上海

法布雷病(FD)是一种罕见的X连锁遗传溶酶体贮积症,因GLA基因变异引起α半乳糖苷酶A(α-Gal A)活性异常,导致代谢底物在多脏器贮积。
法布雷病(FD)是一种罕见的X连锁遗传溶酶体贮积症,因GLA基因变异引起α半乳糖苷酶A(α-Gal A)活性异常,导致代谢底物在多脏器贮积。该病多系统累及和缺乏特异性临床表现的特点,使法布雷病的诊断和鉴别始终充满挑战。
近年来,该病在筛查、诊断、治疗方面不断取得新进展。基于治疗方面的快速发展,国内本领域专家撰写《中国法布雷病诊疗专家共识(2021年版)》(以下简称”本共识“)[1],进一步促进了我国罕见溶酶体病领域的发展。

图1:共识发表于中华内科杂志[1]
被“低估“的罕见病
GLA基因突变导致 α-半乳糖苷酶A(α-GalA)活性部分或全部丧失,造成其代谢底物三己糖酰基鞘脂醇(GL-3)及其衍生物脱乙酰基 GL-3(Lyso-GL-3)在人体各器官及组织内大量贮积,会引起多脏器、多系统损害,可累及肾脏、心脏和神经系统等。
法布雷病在我国《第一批罕见病目录》中排名第27位,长期以来都被视为一种罕见的疾病。据历史区域性流调估算,其综合患病率介于1/117000至1/40000之间,其中男性患病率高于女性。然而,近年来临床对法布雷病的持续了解发展先这一数字可能被低估,如南京对新生儿进行基因筛查的结果显示实际发病率高达1/1321(N=17171)[2]。
此外,对67个高危人群筛查项目(涉及51363名患者)的汇总分析揭示,在左室肥厚(LVH)或肥厚型心肌病(HCM)的患者中,法布雷病的患病率高达0.93%[3]。由此可见,法布雷病的实际患病率可能远高于之前的估计。
同时,多学科专家指出,法布雷病并不少见,甚至可能存在地域性聚集现象。由于该病常累及多个脏器,高危人群及家系的筛查显得尤为关键。患者可能因肾功能异常、胸闷气急等症状就医,其临床表现多样无疑增加了确诊的难度。因此,对于法布雷病的认识和诊疗需要更加深入和全面。
多系统受累,临床表现多样
法布雷病临床表现多样,常涉及神经、肾脏、心脏、皮肤、胃肠道及眼部等多个脏器的受累(图表1)其症状常随着患者年龄的增长而逐步显现,且男性患者的临床症状往往比女性更为严重。
在儿童期,患者可能出现阵发性双足烧灼样疼痛,这种疼痛常因天气变化而诱发。同时,患者还可能伴随出现蛋白尿或在“坐浴区”(如外阴、外生殖器、臀部等部位)的毛细血管角化瘤。这些症状多在青少年时期出现,并随着病程的进展而逐渐加重。
值得注意的是,肾脏、心脏和脑是法布雷病后期的主要受累脏器。因此,相较于普通人群,法布雷病患者罹患心脏、脑血管、肾脏等疾病风险更高。
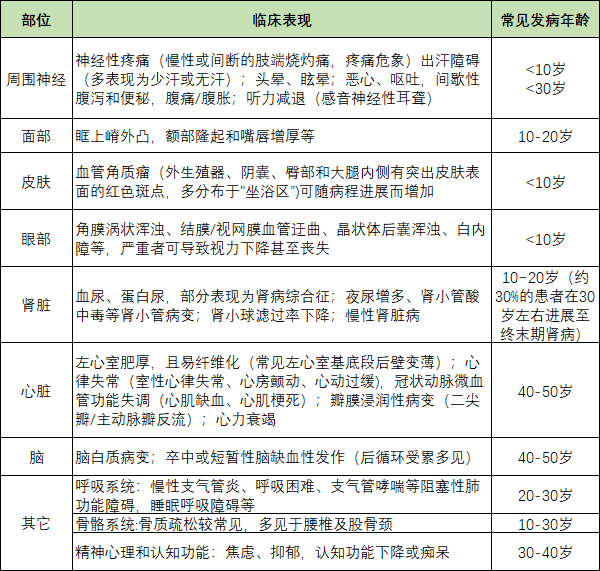
表1:法布雷病受累部位的临床表现[1]
法布雷病按临床表现主要分为经典型和迟发型(如表2)。据国外文献报道,迟发型的发病率较经典型高出10倍。其中,经典型患者以男性为主,其α-半乳糖苷酶活性明显降低甚至完全丧失,导致严重的脏器损害;而迟发型患者则表现为酶活性部分下降,主要影响肾脏和心脏等器官。

表2:法布雷病的临床分型[1]
总之,本共识不仅将法布雷病各受累部位的临床表现与常见发病年龄相对应,还进一步明确了法布雷病的临床分型特征,有助于临床医生规划患者的个性化治疗方案。
早诊断,早干预,多学科管理
目前,法布雷病的诊断面临较高的误诊和漏诊风险。法布雷病的多系统体征和症状不具备特异性,需与多种疾病进行鉴别诊断,同时该疾病进展具有高度可变性。
法布雷病的早期诊断为患者提供在不可逆的器官损伤发生之前进行干预的宝贵机会。除综合考虑临床表现和家族史等关键信息,诊断法布雷病还需借助一系列检测方法,包括α-Gal A活性检测、基因检测、生物标志物检测,活检组织病理学检查等。
◁ α-Gal A活性检测
该法最为简单快捷。广泛用于法布雷病男性疑似患者,男性疑似患者的α-Gal A活性严重下降或缺失,可提示该患者患有法布雷病。女性患者受X染色体随机失活的影响,α-Gal A活性水平不一,缺乏特异性。 需要结合基因检测、底物及衍生物水平来明确诊断。
◁ GLA基因检测
法布雷病诊断的重要检测手段,可提取外周血、干血纸片法(DBS)样本或头发毛囊等组织的DNA,以帮助明确诊断,确定基因变异类型,帮助判断临床表型和指导家系筛查。对意义不明的基因变异(VUS)的解读还需结合家族史、临床表现、底物及衍生物水平、病理等综合判断。
◁ 生物标志物检测
血浆GL-3是诊断法布雷病的重要生化指标,尤其适用于男性患儿。Lyso-GL-3作为更敏感的特异性生物标志物,有助于区分法布雷病的不同类型,对辅助诊断VUS和监测疾病进展具有重要意义。然而,对女性患者的诊断中,Lyso-GL-3的假阳性率仍偏高。
值得一提的是,其中新兴的DBS(干血纸片法)多指标分析也是一种便携、可及的筛查方法,可实现包括酶学、生物标志物和基因三联检测,临床操作简便、快速、准确,可提高法布雷病诊断的准确性。
本版共识新增诊断流程(如图2),为医生提供清晰、易辨的诊断思路,规范诊断流程。需要指出的是,任何一项诊断手段都存在假阳性或假阴性的可能,所以对于临床表现不典型的患者,尤其是女性患儿,建议优先做基因筛查,再结合具体情况参考其他诊断结果综合判断。
组织病理学活检具有辅助诊断意义,可检测受累器官如肾脏、心脏、皮肤或神经组织的病变情况,寻找特异性突变如肾脏病理在光镜下相应组织细胞呈空泡改变,在电镜下相应的组织细胞胞质内充满嗜锇性“髓样小体”。但为有创操作,非必须评估指标。
鉴于法布雷病累及多器官的特点及其治疗现状,我国于2023年8月发布了《法布雷病多学科联合全程管理路径2023》[4],强调多学科团队在法布雷病诊治全程中的重要作用。这一多学科团队涵盖肾内科、心血管内科、神经内科和儿科等,相关医生需熟悉法布雷病的实验室诊断方法,以便及时发现并检测疑似患者。
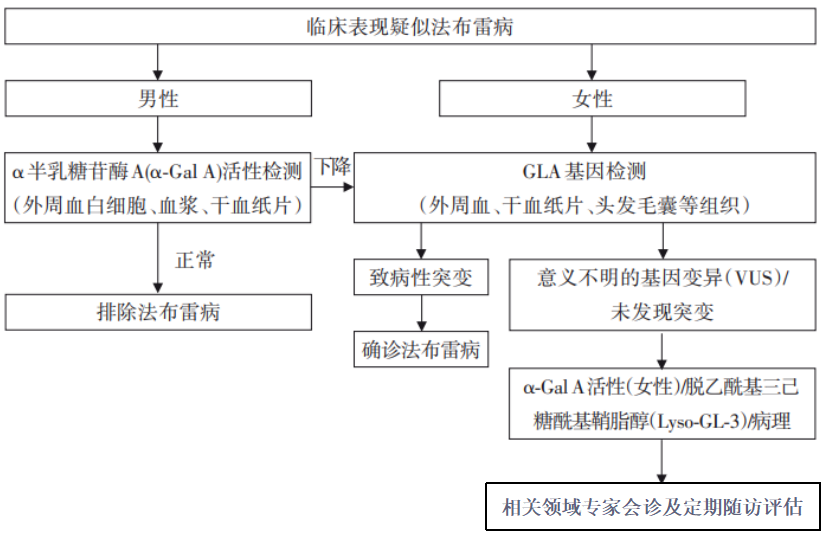
图2:法布雷病诊断流程[1]
搭上酶替代治疗的”早班车“
法布雷病的治疗目标在于延缓疾病进展,改善生活质量,降低相关并发症的发病率,延长患者生存期。因此,在对患者受累脏器初步评估的基础上,制定合适的个体化治疗方案,定期检测和调整治疗,优化患者的疾病管理。
近年来,我国法布雷病领域的最大进展是酶替代治疗(ERT)的进入——该特异性治疗能改善患者预后,从而延缓疾病进展。
ERT主要是通过定期静脉补充人工重组α-GalA,减少细胞内GL-3及Lyso-GL-3贮积,从而达到缓解神经疼痛、改善和延缓心功能衰竭、改善肾小球病变和延缓肾功能不全进展等目的,从而改善患者的生活质量及预后。
2019年12月18日,注射用阿加糖酶-β作为中国首个获批用于治疗法布雷病的ERT药物,成功填补了国内在这一领域的空白,使其成为为数不多的可诊可治的罕见病之一。2020年8月28日,注射用阿加糖酶-α的获批进入中国,为更多法布雷病患者带来了新的治疗选择和希望。
得益于我国政府对罕见病诊疗筛查的大力扶持,法布雷病的ERT治疗已顺利纳入中国医疗保障体系,目前已有部分患者在各地接受规律治疗。此外,本版共识指出,对于有症状的儿童,不论性别,建议立即启动ERT治疗;经典型成年男性患者一经确诊,也应立即启动ERT;其他有症状的患者同样需尽早启动ERT,以最大程度地改善疾病预后。
同时,越来越多的国外指南/共识也表明早期开始ERT的重要性。2016年,美国《儿童法布雷病的管理和治疗:基于美国观点》[5]提出,有症状的儿童患者,不论男女性别,一旦出现FB症状,均应考虑使用ERT治疗;2018年,《加拿大法布雷病治疗指南》[6]进一步明确了法布雷病诊断的评分标准,并在治疗中明确提出不建议随意切换雌激素替代疗法药物。
除了ERT治疗外,还需关注各脏器受累情况,并实施对症辅助治疗(如表3)。同时,新的治疗方法如分子伴侣疗法、基因治疗等,也在投入临床使用或正在研发中,将给患者带来更多的选择与希望。
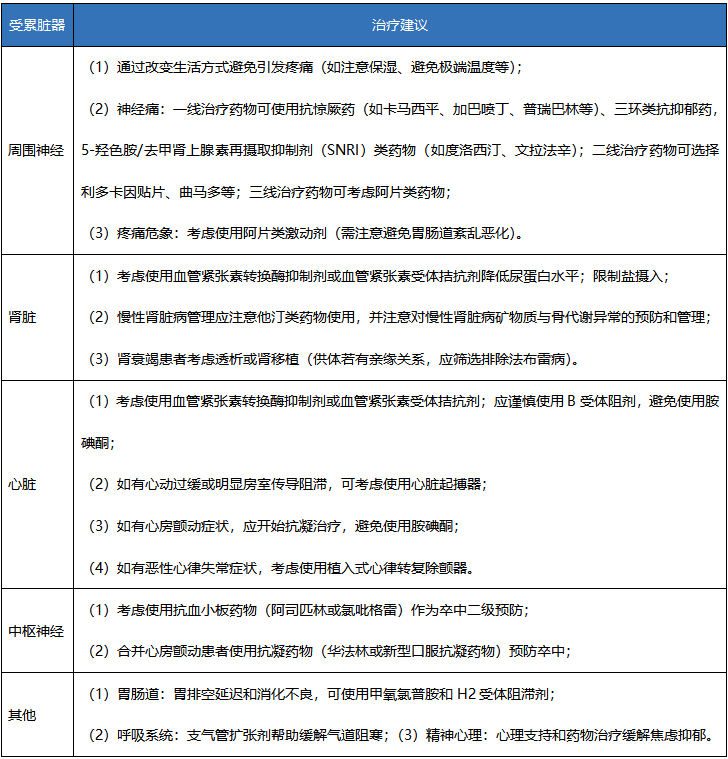
表3:法布雷病对症治疗的建议[1]
注:由于胺碘酮对溶酶体的pH值及酶活性有影响,并已有报道法布雷病患者使用胺碘酮后出现失代偿性心力衰竭,因此建议法布需病患者谨慎使用胺碘酮。
ERT是目前针对法布雷病特异性治疗的基石,已被列入中国、欧洲、美国及其他国家的指南性文件中。对于法布雷病患者来说,搭上ERT的”早班车“,可以获得更好预后。
综上所述,法布雷病累及多系统器官,早筛、早诊、早治是疾病管理的核心。其诊断主要依据酶活性和基因检测,同时辅以底物及衍生物(如GL-3及Lyso-GL-3)检测及病理检查,以提供全面诊断信息。此外,ERT是中国法布雷病儿的唯一特异性治疗手段。且该病的管理需多学科协作,可帮助临床医生尽早识别、诊断法布雷病患者,以及早期给予ERT等治疗,积极改善患者症状及预后。
参考文献:
1.中国法布雷病专家协作组.中国法布雷病诊疗专家共识(2021年版)[J].中华内科杂志, 2021, 60(4):10.DOI:10.3760/cma.j.cn112138-20201218-01028.
2. Yun Sun et al. Newborn genetic screening for Fabry disease: Insights from a retrospective analysis in Nanjing, China. Clin Chim Acta. 2024 Apr 15:557:117889. doi: 10.1016/j.cca.2024.117889. Epub 2024 Mar 24.
3.中华医学会心血管病学分会,中华心血管病杂志编辑委员会.成人法布雷病心肌病诊断与治疗中国专家共识[J].中华心血管病杂志, 2024, 52(02):128-136.DOI:10.3760/cma.j.cn112148-20231008-00263.
4.法布雷病全国专家协作组, 中国医药教育协会临床肾脏病学专业委员会. 法布雷病多学科联合全程管理路径[J]. 中华内科杂志, 2023, 62(8): 949-955. DOI: 10.3760/cma.j.cn112138-20230218-00095.
5.Hopkin RJ, Jefferies JL, Laney DA, et al. The management and treatment of children with Fabry disease: A United States-based perspective[J]. Mol Genet Metab, 2016, 117(2): 104-113.
6.Sirrs S, Bichet D G,, Iwanochko R M, et al. Canadian Fabry disease treatment guidelines 2018[J]. 2020.

如遇到疑似患者,添加微信领取采血包免费安排送检
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



