
让“生物仿制药”最终成为生物仿制药
2025-01-14 小药说药 小药说药 发表于陕西省

2004 年欧洲率先制定生物仿制药法规,受不确定性影响。如今经验表明生物仿制药与原研药质量差异小,监管机构正重新评估指南,这将降低成本,提高生物药可负担性与可及性。
前言
第一部生物仿制药法规和指南规定了专利和营销排他性到期的生物制剂仿制药的营销授权技术要求。该法规由欧洲药品管理局(EMA)于2004年率先制定,不仅使生物仿制药在欧盟(EU)的引入成为可能,而且为全球监管机构树立了榜样。目前,有100多种生物仿制药进入临床使用,生物仿制药的总临床经验超过1500万治疗年。
然而,欧洲的指导方针主要基于不确定性,经过二十年的生物仿制药开发和使用经验,这些不确定性已不复存在。最初人们认为,即使是制造过程中的微小差异也会导致产品质量差异,因此,原研药和生物仿制药之间的物理化学和生物特性差异是不可避免的。在EMA发布的指导文件中,这一立场规定如下:“生物仿制药和参考产品之间总是存在差异”;“即使是最小的差异也可能对安全性和有效性产生临床影响”;“生物仿制药和参考产品之间的质量可比性是减少临床试验数据的先决条件”;“临床数据不能用来证明质量属性的实质性差异”。
这些说法是令人费解的。如果总是存在差异,即使是微小的差异也可能具有临床意义,那么唯一可能的结论是,所有仿制药都需要完整的临床数据。与此同时,一方面,根据监管机构的说法,临床数据不允许用于证明物理化学和生物学比较研究中的差异,另一方面,原研药和候选生物仿制药之间的差异水平可以减少所需的临床试验数据。
在人体内,原研药和生物仿制药的药代动力学行为最好进行比较。然而,以比较的方式研究疗效的概念遇到了临床方法学问题。随着候选生物仿制药的物理化学和生物学相似性的增加,需要提高临床试验检测的灵敏度,以排除安全性和有效性的差异。因此,相似性越大,需要的患者就越多,这导致了一种方法论悖论。
来自已上市生物药的见解
目前,超过25%的新批准药物是生物药。与小分子不同,生物药没有与其代谢相关的毒性。它们的药理学是通过与细胞表面的配体和受体结合,或与自由漂浮在细胞外液中的配体结合来确定的。因此,人源化生物药在任何适应症中的疗效主要取决于其对所结合生物分子的亲和力。
如果制造商能够证明生物仿制药对给定适应症的疗效是基于其与特定同源配体的结合亲和力,并且原研药的结合常数在统计学上与生物仿制药的结合常数没有区别,那么监管机构可能会认为这一证据是必要和充分的,而不需要通过临床试验进行确认。此外,如果原研药的功效是基于其与细胞表面受体结合时向活细胞传递信号的能力,则细胞培养试验同样可用于证明其与原研药的“相似”功效。如果制造商能够证明原研药和生物仿制药在细胞培养试验中通过特定细胞系中的特定受体传递的信号强度是“等效的”,则监管机构可能会认为这一证据足以得出不需要进行疗效试验的结论。
正如生物制药安全指南中说明的那样,生物药的副作用几乎总是它们发挥药效学作用的结果。阿达木单抗和英夫利昔单抗都是治疗类风湿性关节炎、克罗恩病和慢性严重斑块状银屑病等疾病的抗TNF-α抗体,它们阻断TNF-α的生物活性会抑制免疫系统。这会导致副作用,如细菌、病毒或真菌引起的严重感染。用于治疗与慢性肾功能衰竭相关的症状性贫血的依泊汀出类似的安全性和有效性。基于其作用机制,与促红细胞生成素受体结合会刺激红系祖细胞的分裂和分化,从而增加全身血红蛋白和红细胞压积;这会导致血液粘度升高,增加血栓栓塞的风险;贝伐单抗是一种用于治疗各种癌症的抗VEGF抗体,其靶向VEGF-A,VEGF-A是VEGF的一种亚型,可刺激内皮细胞增殖和随后的迁移,从而抑制血管生成以抑制恶性细胞生长和血管形成。同样的药效学也会导致副作用,如高血压和伤口愈合并发症。
在过去的40年里,有报道称上市的生物药存在少量安全问题。最著名的病例是接受阿法依泊汀(Eprex)治疗的患者出现严重的纯红细胞再生障碍性贫血(PRCA),这是由于制剂在批准后发生了多次制造变化,导致产品聚集。另一个案例是在接受干扰素-β1a(Rebif)治疗的患者中意外发生的血栓性微血管病,这与制剂批准后的一次生产变更有关,即从产品组合物中去除白蛋白作为赋形剂。此外,接受不同批次西妥昔单抗(Erbitux)和α-葡萄糖苷酶(Lumizyme)的患者之间的药代动力学暴露差异与Erbitux的不同生产地点(欧盟和美国)以及原料药批次的变化有关,Lumizyme从160升扩大到2000升,导致糖基化谱不同。这些经验表明,原研药也可能因其专有制造工艺的变化而出现问题。
来自生物仿制药的经验
为了进一步了解生物仿制药的安全性和有效性,下表总结了截至2023年8月的欧洲生物仿制药授权公共评估报告(EPAR),并评估了它们与各自原研药的理化相似性,以及比较药代动力学和效果相似性。可以发现,在使用生物仿制药的过程中,除了原研产品中已经发现的问题外,没有发现任何安全性或有效性问题。这表明目前欧盟市场上的所有生物仿制药与原研药的质量差异很小,没有报告任何生物仿制药特有的安全问题。
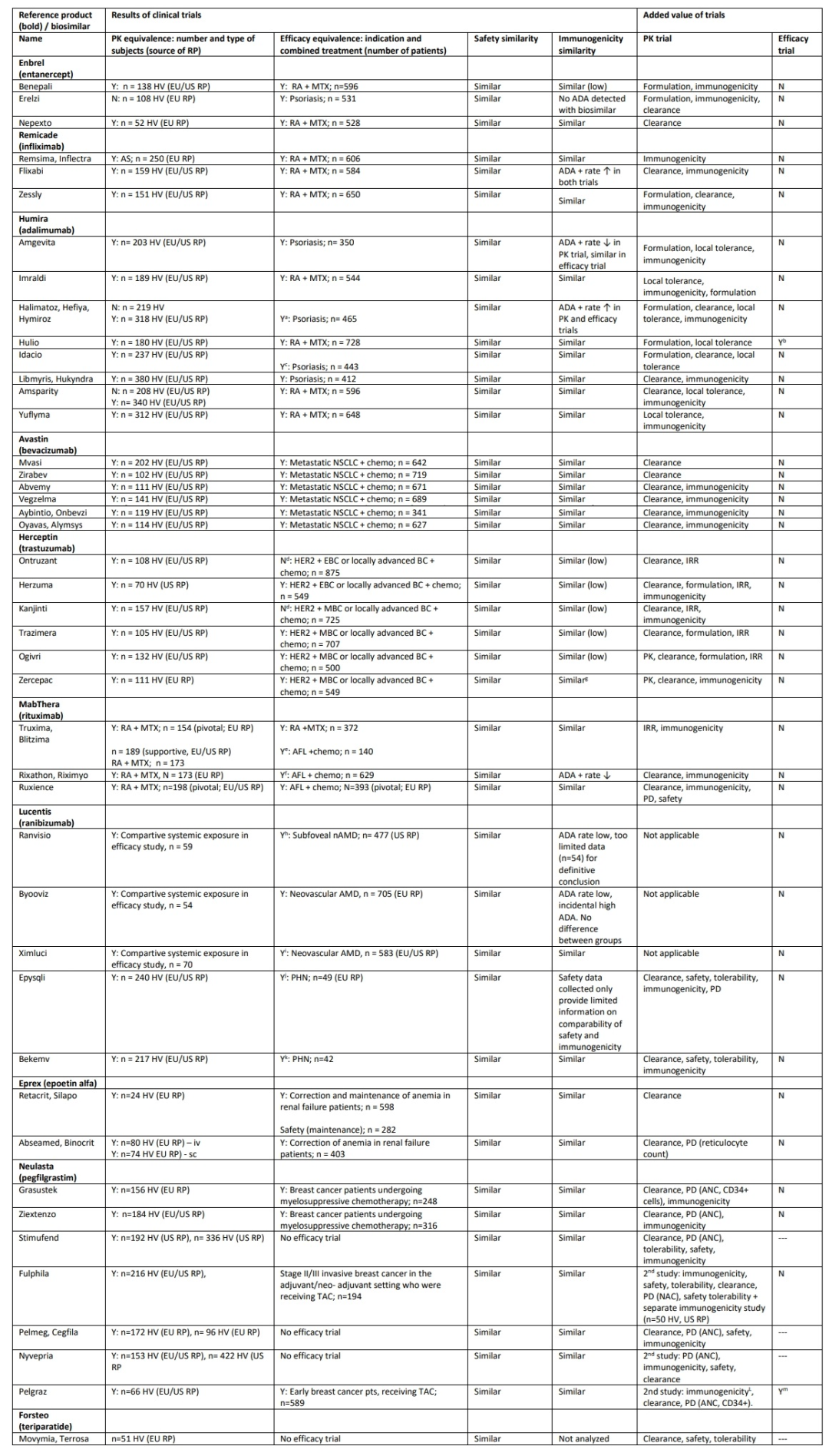
这些生物仿制药的开发及其广泛的临床应用使人们对产品批次之间以及原研药与其生物仿制药之间的差异程度有了更深入的了解。生物药的内在异质性通常反映在抗体、溶酶体储存酶、丝氨酸蛋白酶抑制剂和促红细胞生成素的糖基化中观察到的变化。例如,促红细胞生成素唾液酸化和N-聚糖强烈影响分子半衰期和造血活性;参与溶酶体贮积病的酶的糖基化对其药理学性质很重要。重组变体的糖基化通常是不完全的,例如缺乏甘露糖和N-乙酰半乳糖胺基团的完全唾液酸化,导致与天然对应物相比半衰期更短或内吞作用更差。
由于生物仿制药和原研药批次的广泛比较是任何生物仿制药开发的一部分,一些报告揭示了原研药批次之间的重要批次差异。这些差异有时会超过原研产品和生物仿制药之间允许的差异,但这些差异都不会影响临床安全性和有效性。例如,在曲妥珠单抗的生物仿制药SB3(Ontruzant)的开发过程中,发现了原研药糖基化和效力的质量变化,即抗体依赖性细胞介导的细胞毒性(ADCC)。糖基化和ADCC的漂移与制造地点和工艺的多种变化有关,在上市授权时被认为与临床无关。这一立场后来得到了证实,因为尽管在3期试验的3年随访研究中,受影响的赫赛汀批次的疗效似乎有所降低,但在5年随访中没有观察到疗效降低。
指导方针的调整
目前,监管机构越来越多地考虑到验证性临床试验缺乏敏感性,这也反映在更新的指导文件中,其中PK/PD试验被提议作为具有临床终点的试验的替代方案。EMA、美国食品药品监督管理局(FDA)、英国MHRA和世界卫生组织也在重新评估生物仿制药指南。世界卫生组织和MHRA认识到比较PK/PD临床研究比疗效研究更有价值。目前FDA的指导方针概述了如何在没有比较临床疗效研究的情况下,根据PK和PD生物标志物数据批准生物仿制药。
用更敏感和更小的PK/PD研究取代疗效性研究将有助于在生物仿制药开发中采用更科学的方法。减少临床试验也会降低开发成本,这反过来可能会提高相对昂贵的生物药的可负担性和可及性。此外,生物仿制药指南应考虑到分析技术和生产方法的进步:可以检测到原研药和生物仿制药之间的差异越来越小,制造技术也逐步成熟,生产方法在生物药之间变得高度可比。这些技术进步大大降低了生物仿制药与其原研药之间出现或遗漏具有临床意义的差异的可能性。生物药物法规的精简和合理化将满足社会对负担得起的药物日益增长的需求。换言之:让生物仿制药最终成为生物仿制药。
参考文献:
1.The devolution of biosimilars regulations. Nat Biotechnol.2024 Dec 16.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#生物药# #生物仿制药法规#
23